by: Frithjof Holtz, Merck KGaA, Darmstadt, Germany | Jan 29, 2018
Excipients serve a critical role in the production of final dosage forms for drug products and biologics. They facilitate the manufacturing process (e.g., anticaking agents) and protect, support, and enhance stability. They may also improve bioavailability. In addition, excipients help maintain the safety, or function, of the product during storage and use.
No longer characterized as inert accompaniments to an active pharmaceutical ingredient (API), excipients are the target of an intensified push for more stringent quality management, placing new requirements on both suppliers and users. Regulating excipient quality, however, is no small task. The global market is expected to exceed $5 billion by 2020—with a growth rate of 6.0% from 2014 to 2020 (1). Thousands of different excipients are available, and only a small percentage of them are manufactured solely for pharmaceutical use.
For many years, there have been clearly defined GMP requirements for APIs, including EU GMP Part II, 21 CFR Part 11 and ICH Q7: Good Manufacturing Practice for Active Pharmaceutical Ingredients. But, until recently, well-defined and stringent GMP requirements for excipients did not exist.
A Focus on Quality
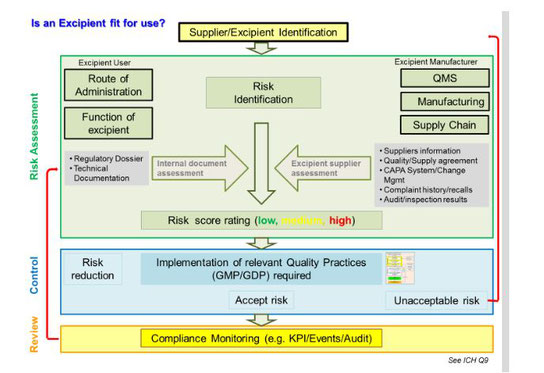
The pharmaceutical industry is increasingly using risk management principles to better protect patients; this renewed focus on safety now includes excipients. At the same time, regulatory authorities have called for more secure supply lines and clearly defined quality measures for excipients.
In 2011, the EU’s Falsified Medicines Directive established that manufacturing authorization holders must use a formalized risk assessment to ascertain the appropriate GMPs for ensuring excipient suitability (2). As part of this risk assessment, manufacturers need to consider both the source and intended use of the excipients in question. The Directive went on to state that the European Commission planned to adopt guidelines for adopting appropriate GMPs for excipients. After robust discussion, guidelines for the risk management process and direction on the appropriate level of GMP for excipients were published in March 2015.
These guidelines apply not only for medicinal products produced in Europe but also for products produced outside Europe intended for the European market. Regulators now expect importers to provide risk assessments and related documents.
As of March 21, 2016, excipient users/ drug product manufacturers in the EU were legally mandated to implement GMP requirements, including completed risk assessments for each excipient used.
Keep in mind, that while regulations regarding GMP for APIs clearly define what is needed for compliance, risk assessment guidelines for excipients are just that—guidelines that offer tools and a framework for determining appropriate GMPs. This leaves the full responsibility of defining what GMPs apply as “necessary” for the excipients of a specific drug product in the hands of the marketing authorization holder.
https://www.pda.org/pda-letter-portal/archives/full-article/formalizing-a-risk-assessment-for-excipients